Co je mitochondriální syndrom? Příčiny, symptomy, terapie, předpovědi
Genetické informace jsou velmi nestabilní. Jedna ze základních principů genetiky říká, že variabilita je hlavním faktorem vývoje všech živých věcí. Mutace jsou nezbytné pro přežití tohoto druhu. Některá variabilita, zejména v mitochondriích, vede k negativním změnám v genetické povaze. To je příčina onemocnění nazývaného mitochondriální syndrom.
Tato onemocnění nejsou tak častá, ale výsledek většiny syndromů mitochondriální DNA je velmi nepříznivý.
Mitochondrie. Jejich funkce v buňce
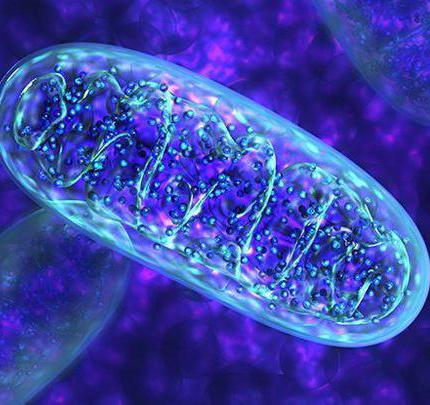
Vzpomeňte si na biologické základy. Mitochondrie je organelie v lidské buňce, která má svůj vlastní kód DNA. Vždy přenášel mitochondriu od matky. Nosí své mateřské vejce. Mitochondrie nezávisle dělí v buňce a opakovaně opakují DNA sadu, která má asi 30 kopií.
Ad
Mitochondriální genom má k dispozici 22 genů pro vlastní transportní RNA; 13 - pro polypeptidy, které jsou obsaženy v supramolekulárních komplexech, které poskytují respiraci organel; 2 geny pro osobní RNA.
Nejdůležitější hodnotou této organely je to, že produkuje ATP. Jednoduše řečeno, je to "elektrárna" v našem těle, bez něho nemohou buňky plně fungovat; rychle "zestárněte" a zemřete.
Co je mitochondriální syndrom?
Když začíná provoz těchto malých "agregátů", začínají problémy s výměnou energie v buňce. U mírnějších forem porušení člověk jednoduše nevydrží fyzické zatížení, které má podle věku tolerovat.
Ad
Závažnější porušení však způsobují nevratné změny v výměně energie a v důsledku toho dochází k vážnému narušení buněk.
Mitochondriální syndrom je komplex chorob spojených s různými vrozenými poškozením mitochondrií.
Příčiny syndromu
Organelles, jako jsou mitochondrie, jsou rozděleny odlišně. Genetická rekombinace není v nich obsažena, ale rychlost mutace je mnohem vyšší. Během rozdělení mitochondrií je distribuce genů mezi novými buňkami zcela náhodná. Pravděpodobnost výskytu mutací je 1 až 99%. A není možné předpovědět.
A čím více nemocných genů, tím větší je pravděpodobnost porušení. Vzhledem k tomu, že mitochondrie jsou dědičná matkou, děti obou pohlaví trpí jejich mutací v jejím těle. A ne selektivně 1 nebo 2. Existuje možnost, že všechny děti budou mít abnormální vývoj orgánů.
Mutace jsou rozděleny do dvou typů. Většina bílkovin je "šifrována" v jaderné DNA, která může být také upravena z nejasných důvodů. Proto sdílejí syndromy způsobené oběma mutacemi běžné mitochondriální kruhové DNA a jaderné.
Symptomatologie
Definování jasné sady symptomů, které jsou vlastnictvím onemocnění, jako je mitochondriální syndrom, je poměrně obtížné. Faktem je, že mutované organely mohou být umístěny v libovolné buňce jakéhokoli orgánu. A čím více se hromadí, tím více narušuje práci tohoto těla a celého systému, ke kterému patří. V mitochondrologii je obvyklé distribuovat syndromy v závislosti na typu postižené tkáně a typu mitochondriální mutace.
Ad
Obvykle jsou postiženy ty orgány a systémy, které nejvíce potřebují konstantní zásobování kyslíkem - to jsou mozek a centrální nervový systém, játra, srdce, svaly. Nedostatek energie, kosterní svaly nepodporují tělo ve vzpřímené poloze. V některých případech se objevují i svalové křeče.
Stává se, že mitochondrie jsou ve své práci tak slabé, že člověk, který dostal takovou sadu organel od matky, je úplně na lůžku. U některých syndromů, které popisují, člověk trpí myoklonem, hepatopatií, záchvaty s časovou demencí a ve velmi mladém věku. Tyto příznaky naznačují syndrom mitochondriálního vyčerpání.
Nejpravděpodobnější abnormality v mitochondriálních mutacích
Celkem bylo zjištěno mnoho forem onemocnění způsobených mutací mitochondrií. Například při ovlivnění svalové kostry jsou epileptické záchvaty diagnostikovány na pozadí nedostatku svalů. Navíc je svalová struktura nejen poškozená, ale pod mikroskopem vypadá jako nerozvinutá červená vlákna. Svalová atrofie v tomto případě se nazývá mitochondriální myopatie. Toto je nejčastější porucha mitochondriálního selhání. Pokud se postihuje srdeční sval - kardiomyopatie, patologické procesy v mozku - zaznamenáme encefalopatii.
Ad
Co je syndrom mitochondriální encefalopatie? Tento syndrom je diagnostikován v případech, kdy dochází k poruchám v genech - tRNA, MTND1, 4-6, MTCYB. To narušuje práci celého nervového systému.
Spolu s encefalopatií je také pozorován příznak, jako je například laktátová acidóza nebo koktem kyseliny mléčné. To je komplikace, kdy kyselina mléčná začne se dostat do krve.
Takové stavy jsou také nebezpečné u pacientů s syndromem mitochondriální insuficience, jako jsou časté a maligní migrény, u dětí dochází ke zpoždění duševního a motorického vývoje, hluchota, ataxie (problémy s rovnováhou).
Příznaky nejsou tak dobře studovány, protože onemocnění spojená s mitochondriemi byla nedávno objevena. Ale řekneme o známých syndromech, jejichž klinické projevy se pokoušejí léčit.
Syndrom MELAS
MELAS (MELAS) - encefalopatie (problémy s centrálním nervovým systémem), laktátová acidóza a navíc mrtvice. Existuje syndrom, jak u kojenců, tak u dospělých. Častěji se příznaky začínají objevovat někde od 5 do 15 let. Jaké jsou tyto příznaky? Jsou uvedeny v názvu syndromu. Pacient najednou zahájí několik úderů v časových a parietálních oblastech mozku. Připojte se k mrtvici a neurologickým problémům. Pak se vyskytuje svalová slabost, smyslová ztráta sluchu. Časté svalové křeče jsou možné.
Příčinou syndromu se považuje náhrada mitochondriálního genu v 3243. pozici. A léčba je možná pouze symptomatická, tj. Podpůrná.
Syndromy delece mitochondriální DNA
Začneme popis s onemocněním, jako je syndrom Kearns-Sayre, který začíná ve věku 4 let. Syndrom se projevuje následovně:
- progresivní oftalmoplegie;
- ataxie;
- atrioventrikulární srdeční blok (zpomalení přenosu impulsů z jedné srdeční komory do druhé);
- retinitis pigmentosa;
- stejná červená trhaná svalová tkáň.
Následující syndrom, který má stejné "kořeny" - Pearsonův syndrom, který se projevuje jinak:
- hypoplastická anémie, první a nejnebezpečnější symptom;
- dysfunkce pankreatu;
- zhoršení zraku je později možné;
- poruchy kostní dřeně;
- vzniku demencí.
Pearsonův syndrom způsobuje delece mitochondriální DNA, stejně jako syndrom Kerns-Sayre. Delece jsou takové změny v sadě chromozomálních genů, v nichž je část genového materiálu zcela ztracena.
Ty alely, které mutovaly nebo ztratily části chromozomů, by se neměly zdát dominantní. Ale v mitochondriální DNA jsou všechny procesy chaotické, mutace se vyskytuje příliš rychle. Někteří vědci dokonce věří, že mitochondrie nejsou organely, ale bakterie, které kdysi vstoupily do lidského těla a zcela se usadily, vytvořily symbiotický vztah s buňkou a začaly sloužit. Tato teorie je naznačena skutečností, že mitochondrie mají svou vlastní, samostatnou kruhovou DNA.
Ad
Bodové mutace
Syndromy způsobené bodovými změnami mateřských mitochondrií zahrnují MERRF syndrom, NAPR, výše zmíněný MELAS a onemocnění, jako je atrofie optického nervu Leber.
MERRF mitochondriální syndrom - jaké jsou jeho vlastnosti?
- Tam je ataxie - porušení koordinace, možná spojené s problémy cerebellum. Člověk má malou kontrolu nad pohyby ve vesmíru.
- Symptomy myoklonické epilepsie.
- Atrofie optického nervu (slepota od narození) a hluchota.
- Laktátová acidóza.
- Zhoršená citlivost.
- Začátek onemocnění nastává ve věku 3 let.

Dalším typem onemocnění, NAPR, je neuropatie, plus ataxie a pigmentosa retinitidy. S tímto syndromem postupuje dítě v psychomotorickém vývoji a demence.
Syndrom deplece DNA
Syndrom vyčerpání mitochondriální DNA je velmi vzácné onemocnění. Dítě s takovým dědičným onemocněním bylo od dětství zakázáno. Tyto syndromy jsou také rozděleny do mnoha druhů.
Mnoho dětí umírá z několika vad ve vývoji vnitřních orgánů předtím, než dosáhnou 3 let věku. Získání takových "zmrzačených" mitochondrií z matky nastává v autozomálně recesivním režimu dědičnosti. Genetika je přesvědčena, že v takových případech dochází k několika vymazáním.
Tento syndrom je také nazýván ve vědeckých kruzích - syndrom mitochondriální deplece DNA. Nemoc se projevuje okamžitě u novorozence. Nemocné dítě má následující vývojové abnormality:
- Silná hepatopatie - porušení jater.
- Vrozená myopatie, vyjádřená výraznou svalovou slabostí.
- Kardiomyopatie - problémy s prací srdce.
- Atrofie svalů a nedostatek reflexů šlach.
Hlavní příčinou těchto onemocnění je vada intergenomického vztahu (komunikace).
Existuje genetika a taková věc, jako je syndrom mitochondriální deplece DNA. Vyčerpání je synonymum pro vyčerpání v genetice. Při tak závažném syndromu je genetický materiál mitochondrií vyčerpaný 70-98%. Poprvé popsáno ne tak dávno, v roce 1991.
Co se stane s dítětem? V novorozeneckém období se již projevuje laktátová acidóza, hypoalbuminémie (prudké snížení albuminu v krvi), edém a těžké selhání jater. Někteří pacienti byli pozorováni a křeče. Příznak, který je viditelný pouhým okem, je silná svalová hypotenze. Všechny děti, které se narodily s takovými označeními, neviděly rok.
Důvod je považován za porušení genu, za který je zodpovědný DNA replikace. Jeho "špatná" práce vede k tomu, že téměř všechny mitochondrie se mění a nevykonávají své funkce. Druh dědičnosti delece mitochondriální DNA může být buď autosomálně recesivní nebo autosomálně dominantní.
Abnormality jaderné DNA
Kromě těchto mitochondriálních syndromů existují i jiné, spojené s abnormalitami jaderné DNA. Tam je také mnoho z nich: Menkes, Leah, Alpers, různé nedostatkové státy. Všichni mají progresivní kurz. Nejnebezpečnější je Leia syndrom, ve kterém dítě prakticky není od narození životaschopné.
Mitochondriální syndrom u dětí
Většina nemocí začíná v raném dětství. Myopatie je převážně převládající, protože děti se nemohou pohybovat samostatně a trpí svalovou bolestí. Kardiomyopatie - dysfunkce myokardu je také běžná.
Mitochondriální syndrom u dítěte, pokud onemocnění nejsou příliš závažné a neohrožují zdraví, po celý život způsobí úzkost a zabrání normálnímu vývoji. Tyto děti potřebují socializační aktivity. Pro ně je důležité vyvinout kosterní svaly, ale ne sportovními metodami (protože mnoho z nich má myokard), ale díky koupání s delfíny. Proto byl pro tyto děti vytvořen speciální fond, kde peníze pocházejí z charity.
 Jednou z forem syndromu mitochondriální deplece DNA je nemocný chlapec pojmenovaný Charlie Guard, narozený v roce 2016. Od narození nemůže spolknout, jíst, dýchat. Jeho stav je zcela kontrolován lékaři a jeho rodiče zoufale bojují o svůj život. Přestože je málo naděje. Má vrozenou hepatopatii, je slepý a má ztrátu sluchu. Jeho rodiče doufají v moderní metody léčby. Tento syndrom získal také "oblíbený" název - Charlieův mitochondriální syndrom.
Jednou z forem syndromu mitochondriální deplece DNA je nemocný chlapec pojmenovaný Charlie Guard, narozený v roce 2016. Od narození nemůže spolknout, jíst, dýchat. Jeho stav je zcela kontrolován lékaři a jeho rodiče zoufale bojují o svůj život. Přestože je málo naděje. Má vrozenou hepatopatii, je slepý a má ztrátu sluchu. Jeho rodiče doufají v moderní metody léčby. Tento syndrom získal také "oblíbený" název - Charlieův mitochondriální syndrom.
Nicméně syndrom mitochondriální deplece DNA jednoznačně vede k smrtelnému výsledku. Lékaři o tom varují rodiče ihned po diagnóze. Vícenásobná léze orgánů a systémů zabraňují normálnímu životu těchto dětí. Proto je pro ženu před těhotenstvím nesmírně důležité podstoupit genetickou analýzu mutací v mitochondriích.
Diagnostické testy
Diagnostika takových syndromů je pro lékaře obtížným úkolem. Při diagnóze je důležitá komplexní analýza různých ukazatelů. Genetická, biochemická, morfologická studie se provádí odděleně, pak se shromáždí všechna data. Dokonce i genealogie dítěte je zkoumána.

Pro správnou lékařskou pomoc je také třeba provést mnoho testů, které měří různé poměry. Například se kontroluje poměr plazmy k laktátu / pyruvátu v krvi. Koneckonců, nedostatek pyruvátu a převaha laktátů může znamenat nástup laktátové acidózy. Je velmi důležité vědět lékaře o poměru ketonových těl v plazmě. Nejúčinnější diagnostickou metodou je svalová biopsie. Forma mutace může být rozpoznána molekulární genetickou analýzou DNA.
Léčba syndromem
Obtížnost léčby spočívá v absenci jakýchkoli mechanismů, které by mohly obnovit "zmutované geny. Lékaři v takových případech nemohou nic dělat, kromě předpisu pyruvátů a některých komplexů vitamínů. Je obzvláště obtížné pomáhat dětem s několika delecemi genů. A pokud karta dítěte naznačuje konečnou fázi syndromu mitochondriální deplece DNA, znamená to, že doktoři úplně podepsali svou impotenci.
Jediná věc, kterou lék může nabídnout, je identifikace mitochondriálních mutací u matky před těhotenstvím. Pak se můžete pokusit jít na mimotělní pojetí, abyste měli zdravé dítě.