Pierre Robinův syndrom: příčiny, léčba, důsledky. Pediatrická maxilofaciální chirurgie
Pierre Robinův syndrom byl pojmenován podle francouzského zubaře, který v roce 1923 objevil spojení ve projevování příznaků této nemoci. Ačkoli v 19. století byly některé z nich popsány v lékařské literatuře. Nemoc je poměrně vzácná, statistiky se však v jednotlivých zemích mírně liší. V Evropě (Dánsku) s výše uvedenou patologií se objevilo 1 dítě z 14 000 novorozenců, které přežily porod a v USA 1 dítě z 20 000 - 30 000 narozených dětí. Výskyt onemocnění je vadou v maxilofaciální oblasti člověka.
Příčiny nemoci
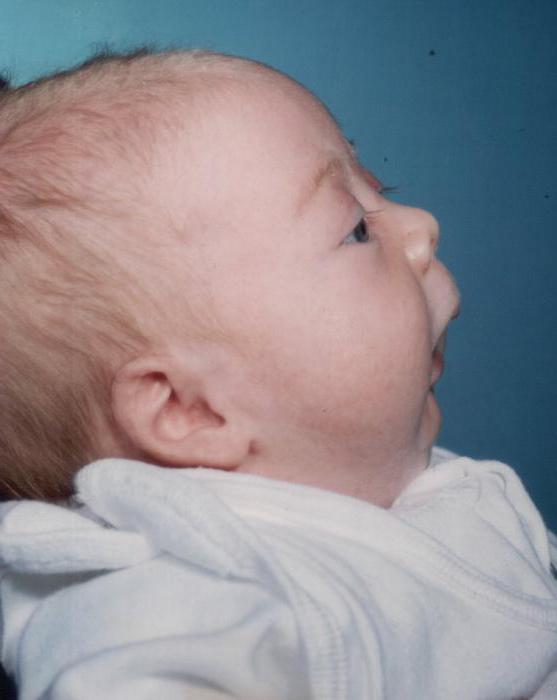
Vývojová porucha spodní čelist a patra se vyskytuje během plodu dítěte. I když nebyly objasněny všechny faktory, kvůli čemuž se objeví syndrom Pierre Robina. Patologie může nastat za různých okolností:
Ad
- V případě infekčního účinku na počáteční fázi těhotenství matky.
- Při nárazu nebo nárazu v děloze během těhotenství.
- U genetických poruch se může objevit také syndrom Pierre Robina (dědičnost vypadá takto - v asi 5% případů se narodí druhé dítě se syndromem, pokud je v rodině jedno nemocné dítě).
- Občas se projevuje patologie, když nese dvě nebo více dětí.
Klinické příznaky

Hlavním příznakem onemocnění je hypoplazie dolní čelisti - její nedostatečné rozvinutí (takové děti mají zkosenou nedostatečně vyvinutou bradu), u které je snížena orální dutina a jazyk je posunut zpět. Na dlaňových deskách dítěte se vytvoří štěrbina. Funkce dýchání a polknutí je obtížné. Velmi často se u Pierre Robinova syndromu objevují další patologie:
Ad
- Porušení struktury sluchadla, které mělo za následek ztrátu sluchu.
- Nemoci centrálního nervového systému (porucha funkce motoru, mikro- a hydrocefalus, zpožděný vývoj řeči a psychika, záchvaty epilepsie).
- Vrozené anomálie vizuální funkce - katarakta, myopie.
- Srdeční onemocnění.
- Přítomnost dalších prstů na končetinách.
- Nedostatek některých končetin.
- Nesprávně utvořený močový systém.
- Porušení tvorby páteře a hrudníku.
- Děti s Pierre Robinovým syndromem se narodily v 20% případů mentální retardace.
U dítěte, které se narodilo s touto patologií, je dýchání často chraplavé a obtížné, sliznice a kůže modrou. Pokud nepomůžete včas, může to být fatální.
Typy nemocí podle obtížnosti

Pierre Robinův syndrom u novorozenců je rozdělen do několika stupňů, v závislosti na schopnosti sebe-aktivitu těla:
- Mírný typ onemocnění (dítě má normální dýchání, dochází k malým potížím s polykáním jídla, které může matka eliminovat).
- Průměrný typ onemocnění (funkce dýchání a spolknutí jsou obtížné, může dojít k asfyxii). Vyžaduje léčbu v nemocniční klinice.
- Závažný typ nemoci (novorozenec sám o sobě je prakticky neschopný dýchání, není možné přijímat jídlo přes ústa). V tomto případě dítě přežívá a vyžaduje okamžitý chirurgický zákrok.
Konzervativní léčba
V počátcích se kojenci, kteří mají v oblasti maxilofaciální poruchy, potopili trubicí a postupně se přeměňovali na kojení. Léčba syndromu má za cíl usnadnit dýchání, uvolnit dítě z nedostatku kyslíku a eliminovat mezeru měkkého patra. Existují případy, kdy se ukáže zlepšení dýchacích funkcí dítěte kvůli pohodlné poloze těla.
Ad
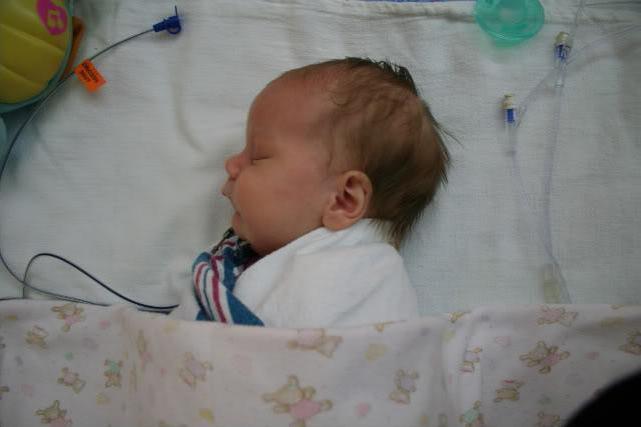
Léčba může být konzervativní a operační, v závislosti na složitosti onemocnění. Ve většině případů, s mírnou závažností, dochází k léčbě umístěním dítěte na břicho. Tato metoda přispívá k růstu spodní čelisti, která posunuje jazyk pacienta dále od zadní stěny, což vede k snadnějšímu dýchání. Když se dítě pohybuje nebo pláče, jeho dýchání se zlepšuje, ale během spánku se opět stává obtížným, proto je nutný neustálý dohled nad takovými dětmi.
Používají se také různé léky - antikonvulzivní a sedativa (fenobarbital, Sibazon a další). Po prvním měsíci života dítěte je vhodné provést neurosonografii (ultrazvukovou diagnostiku mozku), aby zjistila abnormality ve své struktuře.
Operační intervence
Pokud má Pierre Robinův syndrom mírný stupeň závažnosti, aby se zabránilo chirurgickému zásahu, dítě se položí na horní čelist plastovou destičkou, zablokuje rozdělené patra a tlačí jazyk dítěte dopředu.

V závislosti na konkrétním případu onemocnění může lékařský personál provést dočasnou glossopexii (pohyb jazyka od zadní části úst a jeho připevnění k dolnímu rtu dítěte). Toto chirurgie po dobu 1-2 měsíců. Krmení dítěte je v tomto okamžiku zajištěno pomocí gastrostomické trubice (díra, která je vytvořena v žaludku pro krmení dítěte). Gastrostomický výkon se obvykle vytváří ze stěn žaludeční tkáně nebo tenkého střeva. Je vkládán, neustále nebo po dobu krmení, pryžovou trubičku, která slouží jako sonda pro vstup potravy do žaludku.
Ad
Léčba těžkých onemocnění
Moderní maxilofaciální chirurgie zachází se všemi stupni Pierre Robinovy nemoci.
U těžce vyslovené hypoplasie dolní čelisti dítěte se při závažných případech syndromu používá osteosyntéza kompresní distrakční osteosyntézy (chirurgická metoda ovlivnění kostních struktur je také použita k zajištění osteogeneze - obnovení a prodloužení lidských kostí) a tracheostomie (dýchací otvor v průdušce, trubice), aby se obnovilo samo-dýchání.
Pro operaci osteosyntézy jsou vyžadovány speciální prostředky pro fixaci dolní čelisti dítěte, jeho stlačení a prodloužení. Existují zařízení zahraniční a levnější domácí výroby. Přístroj je připojen k dolní čelist dítěte dvěma kolíky na pravé a levé straně. Mezi nimi se kosti rozřezávají a spojují se stlačováním po dobu 5 dnů, aby se vytvořila mladá kosti. 6. den se části čelisti pomalu začnou pohybovat od sebe (asi 1 mm za den). Metoda prodlužování mladé kosti se nazývá rozptýlení .
Ad
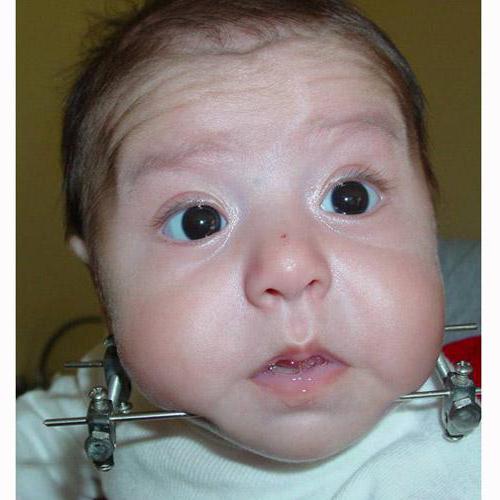
Během operace se používá intubační anestezie (dočasné odpojení citlivosti a vědomí osoby v mozku, paralýza svalových funkcí a zavedení dýchací trubice pro ventilaci plic).
Po 5-6 dnech od začátku prodloužení dolní čelisti dítěte se nejčastěji obnoví nezávislé respirační funkce dítěte. Rozptýlení se provádí, dokud není obnovena správná část obličeje. Na konci tohoto postupu je dítě převedeno do normálního krmení a propuštěno domů. Zařízení jsou odstraněna v celkové anestezii, pouze 3 měsíce po konečné přeměně prodloužené části čelisti do tvrdé kosti.
Odstranění rozštěpeného patra
Operace k odstranění rozštěpu patra je žádoucí provést dříve, než dítě začne mluvit (od 6 měsíců do 1,5 roku). Doba jeho držení závisí převážně na normalizaci dýchání dítěte. Před uranoplastikou se doporučuje nosit zařízení (obturátor), které odděluje nosní a ústní dutiny pro normální dýchání a krmení.
Maxilofaciální chirurgie se obvykle provádí v několika fázích:
- V první fázi se provádí "veloplastika" měkkého patra.
- Ve druhé fázi - plastové tvrdé podnebí. Různí odborníci v oblasti maxilofaciální chirurgie najednou doporučili svůj přístup k době, kdy došlo k odstranění anomálie horního patra. Druhá fáze uranoplastiky může být provedena od 1 roku do 10-11 let.

Důsledky onemocnění
Jak již bylo řečeno, děti, které trpí syndromem Pierre Robina, mohou trochu zaostávat ve vývoji inteligence a řeči, mohou mít problémy s viděním a sluchem a existuje také možnost onemocnění centrálního nervového systému. Ale léčba začala v plné míře a včas může zlepšit nejen vzhled, ale i životně důležitou aktivitu dítěte.