Tay-Sachsova nemoc: symptomy, znaky projevy a léčby
Tay-Sachsova choroba je vzácná genetická patologie, která se vyskytuje jako výsledek mutace genu NHA. Onemocnění bylo objeveno na konci 19. století. Lékařské studie se stále provádějí, jejichž cílem je nalézt lék na toto závažné onemocnění.

Historie objevů
Britský oftalmolog Warner Tay a americký neurolog Bernard Sachs nezávisle popsali onemocnění v roce 1887 a vyvinuli diagnostická kritéria, která odlišují toto onemocnění od jiných neurologických poruch s podobnými příznaky.
Bernard Sachs byl první, kdo předpokládal, že tato patologie je genetická. Jeho intuitivní předpoklad byl potvrzen v polovině dvacátého století po znovuobjevení zákony Mendela.
Bernard Sachs navrhl pro novou chorobu název, který lze nalézt v moderní lékařské literatuře - amarotické rodinné idiocy.
Prevalence
Jak Tay, tak Sachs zaznamenali případy u aškenských židů, mezi nimiž nejčastěji dochází k mutaci genu NEHA. Asi 3% zástupců této etnické skupiny jsou nositeli mutantního genu a výskyt se pohybuje od 1 do 3200 - 3500 novorozenců.
V obecné populaci je každá 300. osoba nositelem Tay-Sachovy nemoci a na 320000 zdravých dětí je 1 pacient.
Příčiny onemocnění
Lékaři už dlouho nemohli odpovědět na otázku, co způsobuje onemocnění Tay-Sachse. Příčiny patologie se staly známými až v polovině dvacátého století, kdy vznikly myšlenky na genetiku. Studie ukázaly, že onemocnění se objevuje v důsledku mutace genu NHA, který se nachází na 15. chromozomu. Onemocnění je typ GM2-gangliosidózy, genetické patologie spojené s nedostatkem nebo sníženou aktivitou hexosaminidázy. Amarotická idiocie je důsledkem poklesu aktivity hexazaminidázy A nebo nedostatečnosti tohoto enzymu.
Onemocnění je přenášeno autosomálně recesivním způsobem, takže jestliže lidský genotyp má zdravý gen HEX, pak neprokazuje Tay-Sachovu chorobu. Genetika onemocnění je podobná dědičnosti takových patologií, jako je Gaucherova choroba, Urbach-Viteova choroba, Dabin-Johnsonův syndrom: jestliže oba rodiče byli nosiči mutovaného genu, pravděpodobnost, že má nemocné dítě, bylo 0,25% a u dětí dochází v téměř 100% případů.

Hlavní formy onemocnění
Obvykle se rozlišují tři hlavní formy onemocnění. Nejběžnější z nich je dítě. Děti s Tay-Sachovou chorobou se normálně rozvinou na 6-7 měsíců. Poté začíná pomalý, ale nezvratný proces duševního a fyzického úpadku.
Existuje také mladistvá forma onemocnění. Ve srovnání s dítětem je méně obvyklé. Až do 3-10 let se dítě vyvíjí stejným způsobem jako jeho rovesníci, ale postupně se začíná pomalý pokles kognitivních a motorických funkcí, dysartrie, dysfágie a ataxie.

Pozdní onemocnění Tay-Sachsovy choroby je nejčastější formou onemocnění. První známky onemocnění se obvykle objevují po 30 letech. Existují však případy a dřívější projevy příznaků (15-18 let). Tato forma onemocnění má nejpříznivější prognózu, protože její progrese může být zastavena.
Symptomy
Bez ohledu na formu nemoci existuje několik hlavních příznaků: dysfagie, ataxie, ztráta kognitivních funkcí, svalová atrofie. Pokud dítě mladší než jeden rok reaguje ostře na drsné zvuky, získává váhu špatně a nedokáže uvolnit svaly, rodiče by to měli ukázat odborníkům - to je to, jak děti začnou Tay-Sachsovu chorobu. Symptomy jsou stále vážnější. Po 6 měsících klesá fyzická aktivita dítě ztratí schopnost sedět samostatně a změnit svou pozici. Slepota se postupně rozvíjí, slyší klesání, atrofuje svaly a rozvíjí paralýzu celého těla.
V mladistvé formě se vedle hlavních příznaků vyskytuje dysartrie (zhoršená řečová jasnost), spasticita, zhoršená koordinace pohybů. Postupně dochází ke ztrátě kognitivních funkcí - ztráta paměti, pozornost, výkonnost. Demence se vyvíjí. V pozdějších stadiích onemocnění se objevují křeče. 
Prvými příznaky onemocnění dospělých jsou onemocnění při polykání, zhoršená koordinace a dysartrie. Často jsou podobné duševní poruchy příznaky schizofrenie (vizuální a sluchová halucinace, apatie, snížená emocionita). Bez léčby je pozorována kognitivní porucha. Pouze pro tuto formu onemocnění existuje účinná léčba k zastavení Tay-Sachsovy nemoci. Národní příručka pro neonatologii říká, že účinné metody pro diagnostiku dospělé formy onemocnění se objevily až v 70. letech předtím, než byla nemoc považována za dětinskou.
Provedení diagnózy
Lékaři se vždy nedaří správně diagnostikovat, pokud jde o tak vzácnou patologii, jakou je Tay-Sachsova choroba. Symptomy, genetika a léčba onemocnění jsou aktivně studovány odborníky. Bez ohledu na formu nemoci existuje několik diagnostických postupů, které se provádějí, pokud máte podezření na její přítomnost. Jedním z nich je stanovení aktivity enzymu hexosaminidázy v séru, leukocytech nebo fibroblastech. U pacientů s Tay-Sachovou chorobou je aktivita hexosaminidázy B vždy pod normou, enzym hexosaminidáza A téměř chybí nebo je její aktivita významně nižší než normální.
Dalším důležitým diagnostickým kritériem je přítomnost jasně červené skvrny na oční rohovce, kterou pomocí oftalmoskopu snadno zaznamenává terapeut nebo oculista. Na všech pacientech byla nalezena červená skvrna na rohovce bez ohledu na věk.
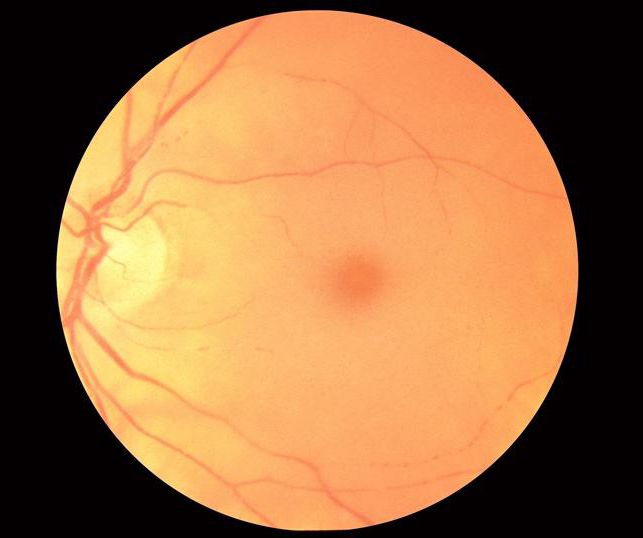
Na rozdíl od jiných onemocnění akumulace lysozomu (Gaucherova choroba, Standhoffův syndrom, nemoci Niemann-Pick), u Tay-Sachsovy choroby nedochází k zvětšení jater a sleziny (hepatosplenomegalie).
Léčba
V současné době neexistují žádné léky, které by dokázaly léčit Tay-Sachsovu chorobu. Symptomy a léčba nemoci jsou stále předmětem vědeckého výzkumu.
Kojenecká forma nemoci Tay-Sachse je nejnebezpečnější. Pokud nemocné dítě nemůže sám spolknout, doporučuje se umělá výživa, není možné obnovit fyzické dovednosti. Žádné drogy, které mohou zastavit nebo zvrátit vývoj nemoci, neexistují navzdory všem snahám vědců. Nemocné děti, i když dostanou nejlepší péči, zřídka žijí ve věku čtyř let.
Pokud je mladistvá forma nemoci důležitá, že dítě je neustále pod dohledem lékaře. Podle pokynů specialisty a procházení všech potřebných lékařských postupů pomůže prodloužit život nemocného dítěte na 12-16 let.
Dospělí forma onemocnění postupuje pomaleji než ostatní a je často léčitelná. Při duševních poruchách se u pacientů předepisuje chlorid lithný nebo cesný. Klinické studie ukázaly, že pyrimethamin může významně zpomalit a ve vzácných případech zcela zastavit progresi onemocnění zvýšením aktivity hexosaminidázy B.

Prenatální diagnostika
Moderní výzkum na počátku těhotenství nám umožňuje zjistit, zda dítě zdědila mutantní gen HEX od rodičů. Pokud jsou oba rodiče nosičem onemocnění, doporučuje se chorionická biopsie. Jedná se o jeden z nejčastějších postupů pro prenatální diagnózu, jehož cílem je identifikovat genetické abnormality plodu. Udržuje se v 10-14 týdnu těhotenství. Amniocentéza také dává jasnou představu o tom, zda je dítě také nosičem mutovaného genu NHA. Tyto postupy mají riziko potratu méně než 1%.
V případě umělé inseminace mohou být genetické abnormality plodu určeny ještě před implantací do dělohy. Pro tento účel se provádí předimplantační genetická diagnostika, analogie prenatální diagnózy. Jeho hlavní výhodou je, že postup není invazivní a naprosto bezpečný. Pouze zdravé embrya mohou být vybrána pro implantaci, čímž se sníží téměř na nulu riziko vzniku nemoci s Tay-Sachovou chorobou.