Marfanův syndrom: znaky, diagnóza, příčiny, příznaky a léčba
Marfanův syndrom se projevuje různými patologiemi kostry, kardiovaskulárního systému, poruchami orgánů zraku. Toto onemocnění je zděděno, je vrozené a pojistné tkáně těla trpí.
Co je první
Marfanův syndrom je porušení normálního fungování pojivové tkáně. Jak se vyvíjí nesprávně, ovlivňuje mnoho vnitřních orgánů. Pacienti mohou mít v těle následující abnormality:
- gigantismus;
- aneuryzma aorty;
- dolichostenomelia a arachnodactyly;
- myopie;
- ektopie objektivu;
- kyfoskolóza;
- deformace hrudní kosti;
- ploché nožky;
- ektázie dura mater;
- acetabulární výčnělek.
Existují tedy běžné fyzikální rysy u lidí, kteří byli diagnostikováni tímto syndromem (Marfanův syndrom). Fotky pacientů ukazují podobnost vnějších příznaků onemocnění a specialisté pochopí do značné míry vzhled pacienta. Mezitím se slabým projevem této nemoci mohou být vnější znaky téměř neviditelné. 
Jak je provedena diagnóza?
Toto onemocnění v lékařské praxi je vzácné. Podle odborníků může mít Marfanův syndrom pouze 10 až 20 tisíc lidí. Diagnostika je založena na několika metodách. Jedná se o rodinnou anamnézu, studie tělesných funkcí, oční vyšetření, rentgenové vyšetření a genetickou prognózu. Současně neexistuje žádný rozdíl, který pohlaví je člověk, na jakou rasu patří a na jakém místě Země žije. Toto onemocnění postihuje jak obyvatele na jihu, tak na severu.
Pokud lékař diagnostikuje Marfanův syndrom, léčba může být konzervativní i chirurgická. V závažných případech je nutné uchýlit se k chirurgickému zákroku, když léčba léky a fyziologickými postupy nedává požadované výsledky.
Chirurgie je často vystavena kardiovaskulárnímu systému a orgánům zraku.
Příčiny onemocnění
Proč se objevuje Marfanův syndrom? Důvody spočívají v mutacích genu FBN1. To je on, kdo je zodpovědný za syntézu fibrillinu. Tento strukturální protein poskytuje pojivové tkáni potřebnou kontraktilitu a elasticitu. S nedostatkem fibrillinu v těle špatně utvořené vláknité struktury. Spojovací tkáň zatímco ztrácí svou sílu a pružnost. Je mnohem horší, dokáže odolat fyziologickému stresu. Od tohoto začátku trpí vnitřní orgány a kostra člověka. 
Kdo nejvíce riskuje
Jak již bylo řečeno, je zděděn Marfanův syndrom, jehož znaky se mohou objevit už v prvních letech života člověka. Pokud někdo v rodině trpěl genetické onemocnění je pravděpodobné, že dítě může také dědictví postihnout.
Ale také se stane, že plod stále má primární mutaci v děloze. V tomto případě je starší žena, tím větší riziko je mít nemocné dítě. To platí zejména u žen, které otěhotní po 35 letech.
Externí projevy
Nejčastěji se tato nemoc projevuje i vnějšími příznaky. Navíc, čím starší je pacient, tím výraznější jsou vnější znaky. U osob, které mají v minulosti syndrom Marfan, je fotka, jak již bylo zmíněno, velmi podobná.
Jejich kostra má jisté rysy - tělo je relativně krátké, vysoké, končetiny jsou dlouhé a tenké, nepřiměřené k kostře. Prsty jsou také protáhlé, pavouci. 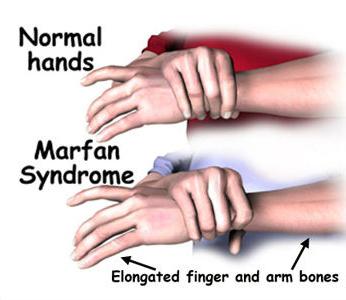
Marfanův syndrom mají příznaky jako:
- astená tělesa;
- nedostatečně vyvinutá podkožní tkáň;
- svalová hypotonie;
- úzká a dlouhá kostra obličeje (dolichocephaly);
- vysoká oblouková obloha, stejně jako porušení kousnutí (prognathia).
Při narození dítěte s Marfanovým syndromem je délka těla u chlapců větší než 53 cm, u dívek - 52,5 cm. Konečná výška je pozorována v oblasti 192 a 175 cm.
Závažnost
Toto onemocnění má různé stupně poškození. Lidé, u kterých je diagnostikován Marfanův syndrom, mohou mít různé příznaky.
Lékaři přidělují několik různých forem této nemoci, v závislosti na tom, jaké životní systémy jsou ovlivněny.
První formulář je smazán. Změny a abnormality ve vývoji jsou mírné. Pouze jeden nebo dva systémy těla trpí a ne moc.
Druhá forma Mafranova syndromu je vyslovována. Existuje zde několik možností:
- Změny jsou mírné, ale existují ve třech systémech těla.
- V jednom systému existují zřejmé odchylky od normy.
- Změny jsou jasně vyjádřeny ve dvou, třech nebo více systémech.
Lékaři také zaznamenají tři závažnosti onemocnění: mírné, středně závažné a závažné. Průběh onemocnění může být také odlišný. Existuje stabilní Marfanův syndrom, jehož známky v průběhu let pozorování zůstávají nezměněny. Existuje progresivní typ onemocnění, když v průběhu času narůstají vývojové patologie a zhoršují se.
Některé rysy této nemoci
Jak jsme již poznamenali, Marfanův syndrom je charakterizován:
- kombinované poškození kostry, očí, kardiovaskulárních a nervových systémů;
- různé fyziologické projevy;
- první příznaky nemoci mohou být diagnostikovány jak při narození, tak po letech;
- chronický progresivní kurz.
U této onemocnění je pozorována dysfunkce kloubů. Odborníci nazývají tuto stavovou hypermobilitu, když jsou klouby snadno překrucovány v různých směrech a velmi mobilní.
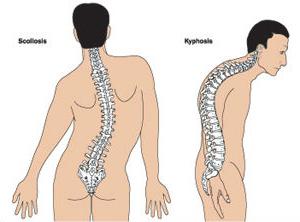
Mnoho pacientů trpících Marfanovým syndromem má nepravidelnou strukturu hrudníku, která má tvar lomu nebo kýlu.
Ortopedové často zaznamenávají deformaci páteře ve svých různých projevech:
- skolióza;
- kyfoskolóza;
- kyfóza;
- dislokace a subluxace cervikální oblasti;
- spondylolistézy.
Pacienti často trpí plochou nohou a protruzí acetabula.
Srdeční komplikace
Kardiovaskulární patologie často dominuje v klinickém obrazu této nemoci. To se projevuje různými způsoby. Pacient může trpět přetrvávajícími vadami v stěnách krevních cév. Ztratily svou pružnost. Aorta, velké větve plicní arterie jsou zvláště náchylné k tomuto. Pacienti mohou mít malformace chlopenního přístroje a srdečních stěn.
U aorty je nejčastěji pozorována postupná expanze její vzestupné části a kroužek ventilu (dilatace, annuloaortic ectasia). Pacient má často aneuryzma, mitrální chlopeň je ovlivněna. Tam je také patologické prodloužení chordae a jejich prasknutí.
U dítěte, které je ještě v děloze, ale na úrovni genu, již byl mu předán Marfanův syndrom, často se tvoří vrozené srdeční vady. Mohou to být:
- koarktaci aorty ;
- defekt síňového septa (ASD);
- plicní stenóza;
- defekt interventrikulární septum (VSD).
Mohou se objevit také poruchy srdečního rytmu (tachykardie, fibrilace síní), vývoj infekční endokarditidy.
V nejnepříznivější novorozenecké formě Marfanova syndromu se srdeční selhání rychle rozvíjí od samotného narození dítěte. Často tyto děti nežijí do jednoho věku.
Zhoršená vizuální funkce
Pacienti s touto nemocí ve většině případů trpí patologií zraku. Mohou to být:
- myopie;
- zploštění a zvětšení velikosti rohovky;
- dislokace / subluxace čočky;
- hypoplázie duhovky a ciliárního svalu;
- změna kalibru retinálních cév;
- strabismus.

Současně ve věku 4 let se ektopie objektivu často rozvíjí, postupuje rychle, zhoršuje se vizuální funkce.
Hlavní léčba
Stojí za zmínku, že s touto nemocí jeden lékař nemůže předepsat léčbu. Odborníci z jiného profilu provádějí pozorování a dávají doporučení: oftalmolog, ortopedista, kardiolog, kardiolog, terapeut, genetik.
Za prvé, léčba je zaměřena na prevenci progrese této nemoci. Vzhledem k tomu, že je konečně nemožné zbavit se nemoci, je důležité jej zastavit, aby v budoucnosti nebyly ovlivněny životně důležité orgány.

Pokud dojde k selhání ventilů srdce, zobrazí se operace. Naléhavě je třeba umístit protézovou mitrální ventil.
Těhotné ženy, jejichž Marfanův syndrom má výraznou kardiovaskulární patologii, jsou označovány za raný císařský řez.
Pokud je zraková porucha, odborníci si vybírají speciální brýle a kontaktní čočky. Se zřejmými patologiemi, kdy jsou diagnostikovány, jako je katarakta, glaukom, je proveden posun čočky, předepisuje se chirurgická nebo laserová léčba.
Operace se také zobrazuje, pokud v kosterním systému dochází k výrazným porušením. Lékaři drží spinální stabilizaci, torakoplastiku, kyčelní artroplastiku.
Kolik pacientů žije s touto chorobou
U Marfanova syndromu je pravděpodobnost náhlé smrti vysoká. Pouze odborníci mohou předpovědět, jak dlouho bude člověk s touto chorobou žít, vzhledem k stupni poškození těla. Včasná léčba a korekce srdeční chirurgie mohou zlepšit kvalitu života a prodloužit její délku na 60-70 let. Nemělo by se zapomínat, že lidé s Marfanovým syndromem musí podstoupit diagnózu a být neustále sledováni lékařem.
Lékaři omezují fyzickou aktivitu na pacienty s tímto onemocněním, jsou kontraindikovány při vysokém zatížení, potápění, kontaktním sportu.
Ženy, které mají v minulosti tuto nemoc a plánovaly mít děti, by měly vyšetřovat genetici.